
Nature | gnomAD数据库研究新成果:基于7.6万人WGS数据构建全基因组突变约束图谱
2023-12-21 测序中国 测序中国 发表于上海

该研究为理解非编码区域的约束提供了新视角;Gnocchi图谱提高了人们对基因组功能遗传变异的识别及理解。
导读
非编码DNA是理解人类基因调控和复杂疾病的核心,研究人员猜测许多致病的基因突变可能存在于调控基因表达的非编码区域中。但系统地识别该区域的有害突变一直是一个挑战。
随着人们对基因的不断探索以及相关方法技术不断发展,由纯化自然选择(约束)引起的破坏性变异的减少已被广泛用于研究人类疾病背后的蛋白质编码基因。2020年,全球多国研究人员合作发布基因组聚合数据库(gnomAD(v2)),其基于人类参考基因组GRCh37,整合了来自15,708个全基因组和125,748个外显子组的预测功能丧失变异,为更广泛的科学研究提供了汇总数据。但目前评估非编码区域约束仍存在困难。

近日,美国Broad研究所领导的研究团队在Nature上发表了题为"A genomic mutational constraint map using variation in 76,156 human genomes"的文章。研究团队聚合并重新处理了gnomAD中的153,030个全基因组,将其与人类参考基因组GRCh38进行比对,保留了来自76,156个个体的全基因组数据,进一步开发了gnomAD(v3),构建了人类全基因组突变约束图谱(Gnocchi),以识别在突变时最有可能导致疾病的非编码区域。此外,研究团队还提出了一个改进的突变模型,通过整合局部序列背景和区域基因组特征以检测变异的减少。综上,该研究为理解非编码区域的约束提供了新视角;Gnocchi图谱提高了人们对基因组功能遗传变异的识别及理解。

文章发表在Nature上
研究团队整合了来自多个种族、健康无相关的76,156个人类全基因组数据,通过严格的质量控制,识别了约6.44亿个高置信度的短单核苷酸变异(SNVs和InDels),这些变异数据的广度和深度为构建Gnocchi提供了坚实的基础。除全基因组数据更多外,该研究还代表了多样的遗传背景,更具祖先多样性,这增加了研究结果的普遍性和准确性。
gnomAD等大型数据集帮助科研人员开发了“不耐受”指标,以确定哪些基因对遗传变异不耐受,并不断将这一指标扩展到人类基因组的非编码部分。蛋白质编码基因有明确的边界,但非编码区域并不能容易地划分为功能单元。
为解决上述问题,Gnocchi将基因组划分为大小为1kb的连续、不重叠的窗口(bins),通过比较观察到的变化和预期的变化并结合改良的突变模型来量化每个窗口的约束程度(图1)。该突变模型结合了局部DNA序列背景、区域基因组特征和甲基化水平,能更准确地评估自然选择对于任一特定DNA区域发生突变的约束程度。
结果显示,Gnocchi能够精准地量化和识别基因组中受到强烈约束的区域;并能准确区分非编码基因组中的良性变异和经筛选的致病突变。Gnocchi为理解基因组中各区域的功能及其在疾病发展中的作用提供了新的视角。
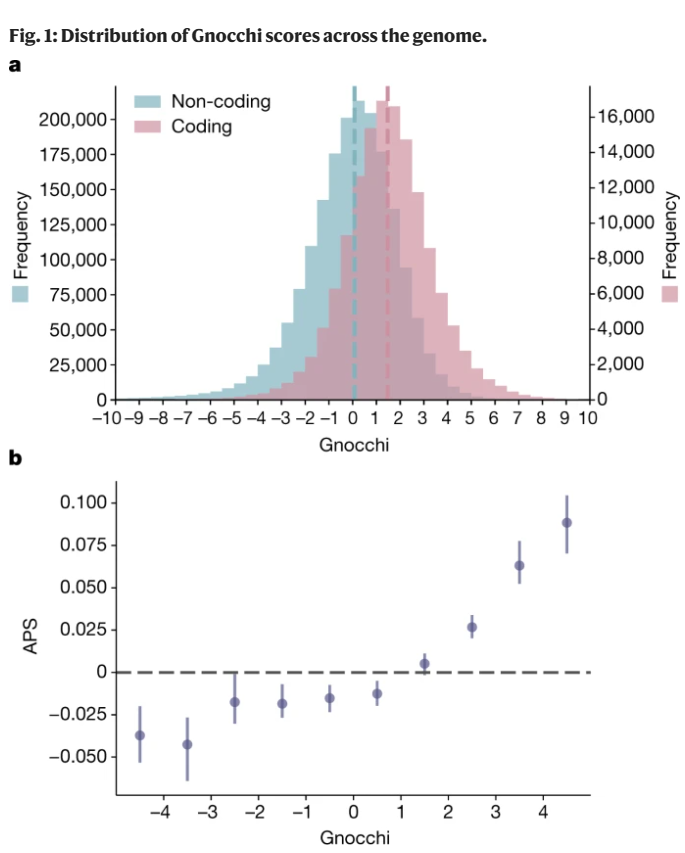
图1. Gnocchi评分在基因组中的分布。
此外,研究团队分析了Gnocchi在识别非编码区域的功能相关性方面的能力(图2),例如顺式调控元件(cCREs)在Gnocchi评分最高的基因组区域中富集,表明这一区域可能在调控关键基因表达中具有重要作用。
分析显示,蛋白编码序列的平均约束强于非编码区域;基因组中的非编码约束区域富含与人类特征和疾病相关的已知调控元件和变异;变异约束的调控元件更倾向于调控那些同样受约束的蛋白质编码基因,这为定位和表征功能性非编码区域提供了有效帮助,能更深入了解相关基因调控机制和网络,及其变异对于人类复杂遗传疾病和表型的影响。
以上研究结果凸显了非编码区域在基因调控中的重要性,也为基因调控机制相关研究提供了新的技术和方向。
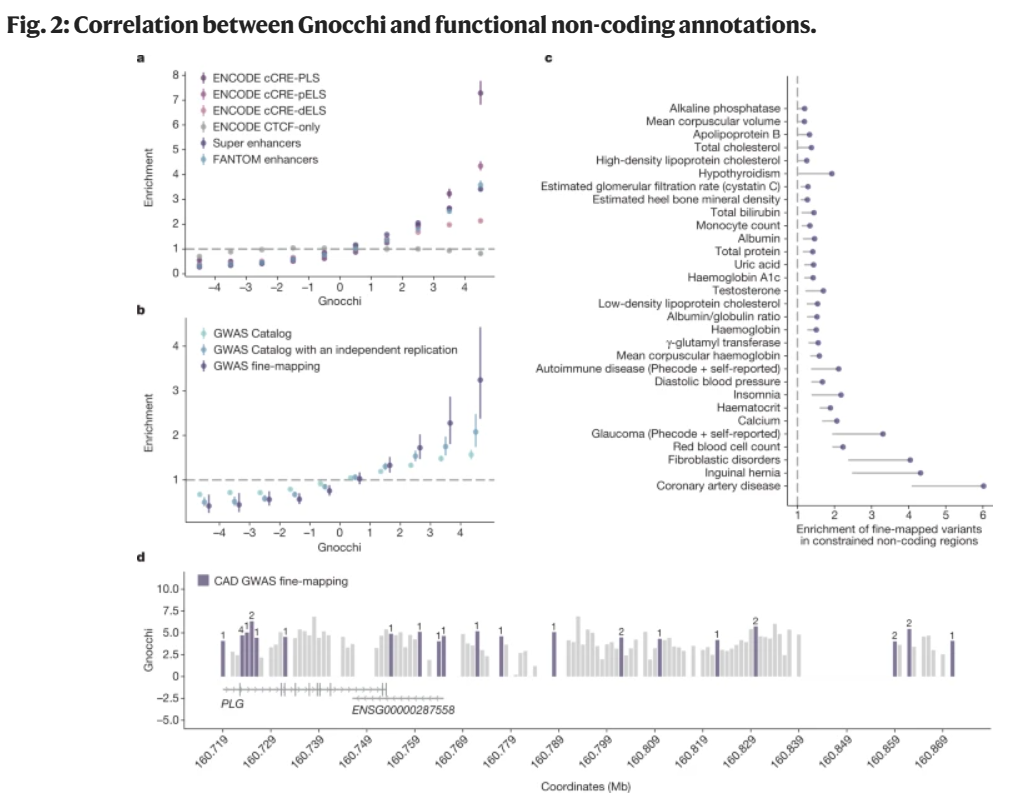
图2. Gnocchi与功能性非编码注释之间的相关性。
Gnocchi的主要进步在于其能够计算每个窗口中变异的理论预期数量。由于局部序列背景和DNA甲基化修饰等因素的影响,基因组的突变率变化较大,依托改良的突变统计模型Gnocchi能够更好地估计每个窗口中的突变率(图3)。与其他非编码约束评估方法( Orion、CDTS和gwRVIS)相比,Gnocchi在识别功能性非编码变异方面表现出一致的高性能,可优先排序非编码变异,揭示了其在基因组研究中的优势和潜力,这对未来的疾病相关基因研究和药物发现尤为重要。
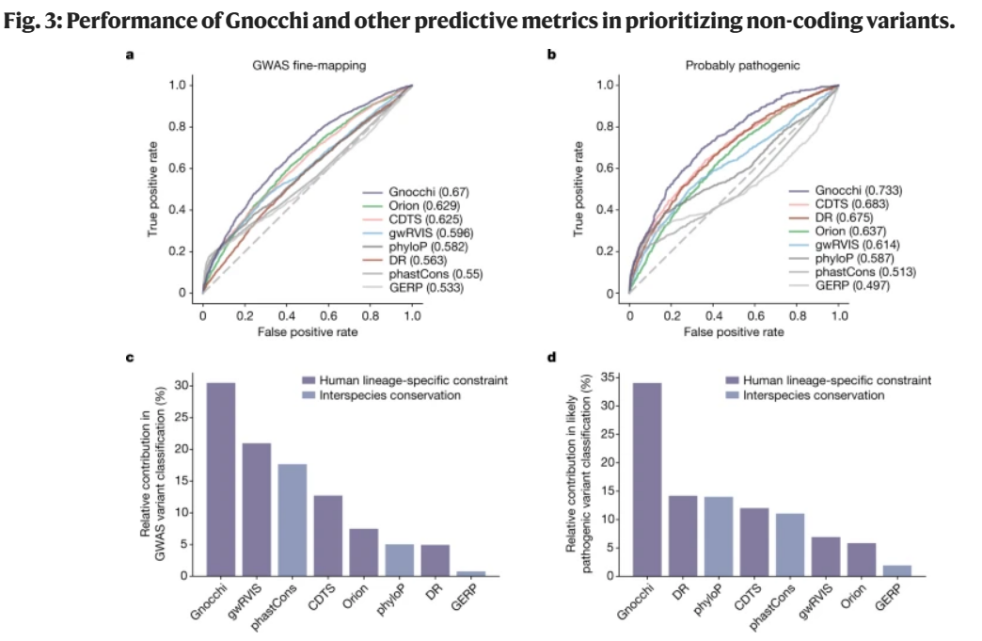
图3. Gnocchi和其他预测指标在对非编码变量进行优先排序时的性能。
除了单核苷酸变异,Gnocchi还强调了拷贝数变异(CNVs)在人类疾病风险变异中的重要性(图4)。分析表明,在发育延迟的个体中,基因组非编码约束区域的CNVs显著过量,这一发现提示Gnocchi可作为受大型CNVs影响的关键区域的有用指标,有助于发现CNVs疾病相关性研究中非编码区域的风险因素。
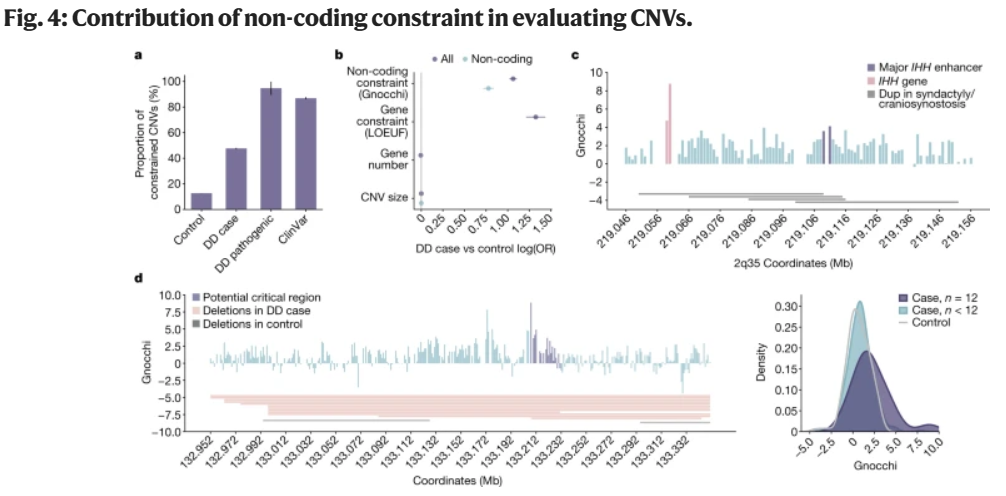
图4.非编码约束对CNVs评估的贡献。
最后,研究团队展示了Gnocchi可以用于提高传统的基因水平不耐受评估方法性能(图5)。研究团队比较了一个基因的非编码增强子对变异的不耐受性(使用Gnocchi检测),与该基因的编码区域对破坏其正常功能的变异的不耐受性(使用LOEUF检测)。结果显示,这两个指标大体上是一致的,但在某些情况下,一个看似对功能丧失有耐受性的基因可能存在一个不耐受的增强子。因此,将一个基因的LOEUF评分与其增强子的Gnocchi评分结合,可以改善小蛋白质编码基因的整体不耐受性评估。
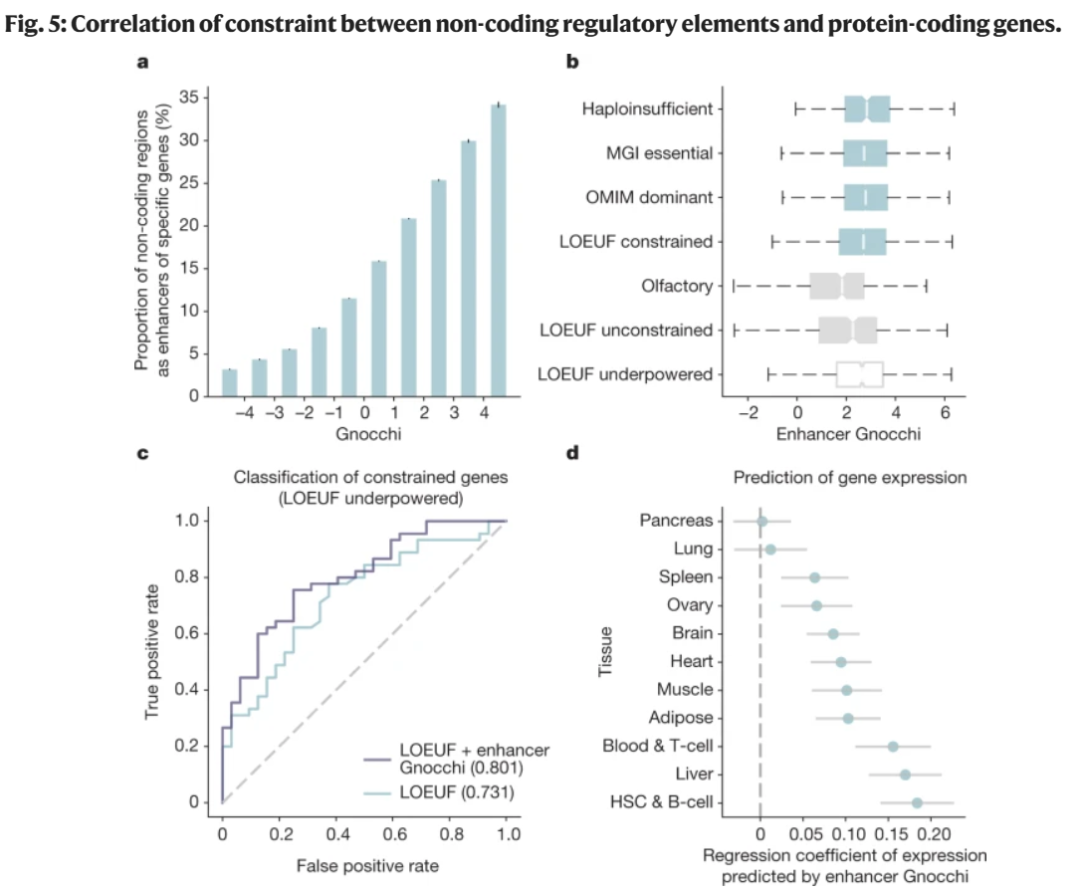
图 5.非编码调控元件与蛋白质编码基因约束的相关性研究。
综上所述,人类全基因组约束图谱的开发是基因组研究领域的一个重要突破,该研究证明了Gnocchi在表征非编码区和蛋白质编码基因方面的价值,并强调了将基因组学的最新成果应用于疾病研究和治疗的重要性,也为探索人类遗传多样性和复杂性提供了新工具。未来,gnomAD将会继续不断扩大数据库,使其更全面地涵盖全球人口的数据样本,并有望揭示人类基因组的隐藏秘密。
gnomAD数据库:https://gnomad.broadinstitute.org/
论文原文:
Chen, S., Francioli, L.C., Goodrich, J.K. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature (2023).
https://doi.org/10.1038/s41586-023-06045-0
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#全基因组# #非编码DNA# #gnomAD# #人类全基因组突变约束图谱#
38