
苯丙酮尿症:症状与体征、病因、流行病学、诊断和治疗
2022-08-20 MedSci原创 MedSci原创

苯丙酮尿症(phenylketonuria,PKU)是一种常见的氨基酸代谢病,是由于苯丙氨酸(PA)代谢途径中的酶缺陷,使得苯丙氨酸不能转变成为酪氨酸,导致苯丙氨酸及其酮酸蓄积,并从尿中大量排出。本病
苯丙酮尿症(phenylketonuria,PKU)是一种常见的氨基酸代谢病,是由于苯丙氨酸(PA)代谢途径中的酶缺陷,使得苯丙氨酸不能转变成为酪氨酸,导致苯丙氨酸及其酮酸蓄积,并从尿中大量排出。本病在遗传性氨基酸代谢缺陷疾病中比较常见,其遗传方式为常染色体隐性遗传。临床表现不均一,主要临床特征为智力低下、精神神经症状、湿疹、皮肤抓痕征及色素脱失和鼠气味等、脑电图异常。如果能得到早期诊断和早期治疗,则前述临床表现可不发生,智力正常,脑电图异常也可得到恢复。2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,苯丙酮尿症被收录其中。
一、一般概述
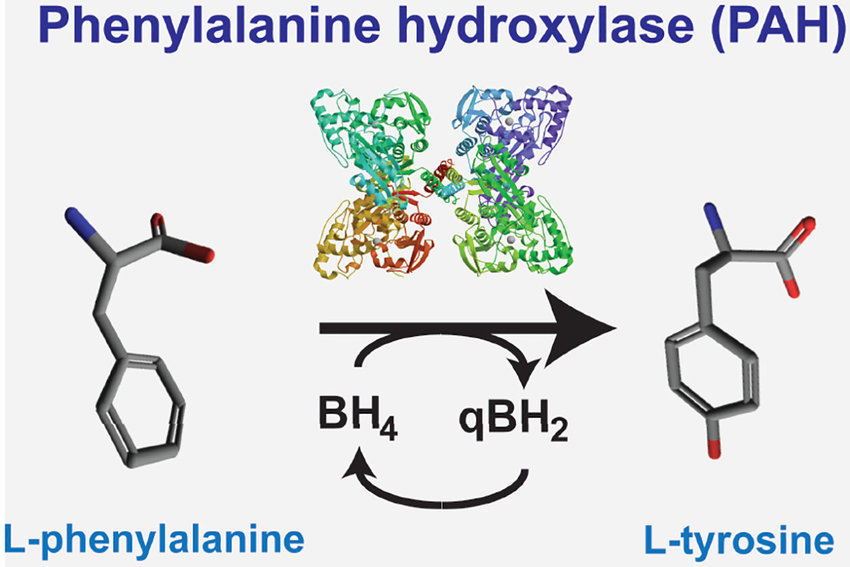
苯丙酮尿症 (PKU) 是一种先天性代谢错误,可在出生后的最初几天通过常规新生儿筛查检测到。 PKU 的特征是缺乏或缺乏一种称为苯丙氨酸羟化酶 (PAH) 的酶,该酶负责加工氨基酸苯丙氨酸。 氨基酸是蛋白质的化学组成部分,对于正常的生长和发育至关重要。 在 PAH 活性正常的情况下,苯丙氨酸会转化为另一种氨基酸酪氨酸。 然而,当多环芳烃不存在或缺乏时,苯丙氨酸会积聚并对大脑产生毒性。 如果不进行治疗,大多数 PKU 患者会出现严重的智力障碍。 为预防智力障碍,治疗包括在生命的最初几天或几周内开始严格控制、限制苯丙氨酸的饮食。
二、症状与体征
1.生长发育迟缓
患有 PKU 的婴儿通常在出生时表现正常。通过早期筛查和饮食治疗,受影响的个体可能永远不会出现 PKU 症状。然而,未经治疗的新生儿在出生后最初几天没有被诊断出来,可能会很虚弱并且喂养不良。其他症状可能包括呕吐、易怒和/或带有小丘疹的红色皮疹。几个月大时发育迟缓可能很明显。未经治疗的儿童的平均智商通常低于 50。PKU 的智力障碍是大脑中苯丙氨酸水平升高的直接结果,这会导致单个神经纤维的脂肪覆盖层(髓磷脂)遭到破坏。它还可以通过降低大脑中多巴胺和血清素(神经递质)的水平而导致抑郁。
2.皮肤毛发表现
未经治疗的 PKU 婴儿的眼睛、皮肤和头发颜色往往异常浅,因为高苯丙氨酸水平会干扰黑色素的产生,黑色素是一种导致色素沉着的物质。它们还可能有一种由尿液或汗液中的苯乙酸引起的霉味或“老鼠味”体味。皮肤常干燥,易有湿疹和皮肤划痕症。
3.神经精神表现
一些未经治疗的 PKU 患者会出现神经系统症状,包括癫痫发作、肌肉运动异常、肌肉紧绷、反射增强、不自主运动或震颤。
4 其它
未经治疗的 PKU 女性怀孕后流产或胎儿生长出现问题(宫内发育迟缓)的风险很高。未经治疗的 PKU 女性的孩子可能有异常小的头部(小头畸形)、先天性心脏病、发育异常或面部异常。这些症状的严重程度与母亲体内高水平的苯丙氨酸之间存在密切关系。因此,所有停止治疗的 PKU 女性都应在受孕前恢复治疗,并在整个怀孕期间继续治疗,由代谢遗传学家和营养师管理。
三、病因
苯丙氨酸是人体必需的氨基酸之一。正常人每日需要的摄入量约为200~500毫克,其中1/3供合成蛋白,2/3则通过肝细胞中苯丙氨酸羟化酶(PAH)的转化为酪氨酸,以合成甲状腺素、肾上腺素和黑色素等。苯丙氨酸转化为酪氨酸的过程中,除需PAH外,还必须有四氢生物蝶呤(BH4)作为辅酶参与。基因突变有可能造成相关酶的活性缺陷,致使苯丙氨酸发生异常累积。见:Neurology:苯丙氨酸对苯丙酮尿症患者脑功能的影响
PKU 以常染色体隐性遗传模式遗传。 当一个人从每个父母那里继承了一个异常基因时,就会发生隐性遗传疾病。 如果一个人接受一个正常基因拷贝和一个异常基因拷贝,他们将成为该病的携带者,但不会出现症状。 两个携带者父母都通过异常基因并因此生育受影响的孩子的风险是每次怀孕的 25%。 男性和女性的风险相同。
已鉴定出 PKU 基因中超过 300 种不同的变化(突变)。 由于不同的突变会导致不同程度的 PAH 酶活性,从而导致血液中苯丙氨酸升高的程度不同,因此必须根据个体特定的苯丙氨酸耐受性来调整每个孩子的饮食。
四、流行病学
据报道,新生儿筛查项目中 PKU 的发病率在美国每 13,500 至 19,000 名新生儿中有 1 名。 PKU 影响大多数种族背景的人,尽管在非洲裔美国人和德系犹太人血统中很少见。
五、检查
1.新生儿期筛查
新生儿喂奶3日后,采集足跟末梢血,吸收再生厚滤纸上,晾干后邮寄到筛查中心,采用Guthrie细菌生长抑制试验半定量测定,其原理是苯丙氨酸能促进已被抑制的枯草杆菌重新生长,以生长圈的范围测定血中苯病氨酸的含量,亦可在苯丙氨酸脱氢酶的作用下进行比色定量测定,其假阳性率较低。当苯丙氨酸含量>0.24mmol/L(4mg/dl)即两倍于正常参考值时,应复查或采静脉血定量测定苯丙氨酸和酪氨酸。正常人苯丙氨酸浓度为0.06~0.18mmol/L(1~3mg/dl)而患儿血浆苯丙氨酸可高达1.2mmol/L(20mg/dl)以上,且血中酪氨酸正常或稍低。
2.尿三氯化铁试验
用于较大婴儿和儿童的筛查。将三氯化铁滴入尿液,如立即出现绿色反应,则为阳性,表明尿中苯丙氨酸浓度增高。此外,二硝基苯肼试验也可以测尿中苯丙氨酸,黄色沉淀为阳性。
3.血浆氨基酸分析和尿液有机酸分析
可为本病提供生化诊断依据,同时,也可鉴别其他的氨基酸、有机酸代谢病。
4.尿蝶呤分析
应用高压液相层析测定尿液中新蝶呤和生物蝶呤的含量,用以鉴别各型PKU。典型PKU患儿尿中蝶呤总排出量增高,新蝶呤与生物蝶呤比值正常。DHPR缺乏的患儿蝶呤总排出量增加,四氢生物蝶呤减少,6-PTS缺乏的患儿则新蝶呤排出量增加,其与生物蝶呤的比值增高,GTP-CH缺乏的患儿其蝶呤总排出量减少。
5.酶学诊断
PAH仅存在于肝细胞,需经肝活检测定,不适用于临床诊断。其他3种酶的活性可采用外周血中红、白细胞或皮肤成纤维细胞测定
6.DNA分析
近年来广泛用于PKU诊断,杂合子检出的产前诊断。但由于基因的多态性,分析结果务须谨慎。
7.其他辅助检查
五、治疗
PKU 的治疗目标是将血浆苯丙氨酸水平保持在 120-360 umol/L (2-6 mg/dL) 范围内。这通常是通过仔细计划和监控饮食来实现的。限制儿童苯丙氨酸的摄入量必须谨慎,因为它是一种必需氨基酸。精心维护的饮食可以预防智力残疾以及神经、行为和皮肤病问题。治疗必须在很小的时候开始,否则可能会出现某种程度的智力障碍。但是,即使是一些晚期治疗的孩子也做得很好。研究一再证明,在三个月前接受低苯丙氨酸饮食治疗的 PKU 患儿表现良好,智商在正常范围内。
如果 PKU 患者停止控制苯丙氨酸的饮食摄入量,通常会发生神经系统变化。智商可能会下降。一旦停止饮食调节,可能出现并变得严重的其他问题包括上学困难、行为问题、情绪变化、视觉运动协调性差、记忆力差、解决问题的能力差、疲劳、震颤、注意力不集中和抑郁。
经过多年的争论,现在临床医生几乎普遍接受这种饮食需要无限期地持续下去,并且在儿童期或以后停止饮食的 PKU 成年人应该恢复饮食。许多年轻人已经重新开始饮食,并发现由于血液苯丙氨酸水平降低,精神清晰度有所改善。
由于苯丙氨酸几乎存在于所有天然蛋白质中,因此不可能在不损害健康的情况下仅使用天然食物充分限制饮食。出于这个原因,特殊的不含苯丙氨酸的食物制剂很有帮助。饮食中通常不允许含有高蛋白质的食物,例如肉、牛奶、鱼和奶酪。水果、蔬菜和一些谷类食品等天然低蛋白食物的数量是有限的。
1.低苯丙氨酸饮食
主要适用于典型PKU以及血苯丙氨酸持续高于1.22mmol/L(20mg/dl)的患者。由于苯丙氨酸是合成蛋白质的必需氨基酸,完全缺乏时亦可导致神经系统损害,因此对婴儿可喂给特制的低苯丙氨酸奶粉,到幼儿期添加辅食时应以淀粉类、蔬菜、水果等低蛋白食物为主。苯丙氨酸需要量,2个月以内约需50~70mg/(kg·d),3~6个月约40mg/(kg·d),2岁约为25~30mg/(kg·d),4岁以上约10~30mg/(kg·d),以能维持血中苯丙氨酸浓度在0.12~0.6mmol/L(2~10mg/dl)为宜。饮食控制至少需持续到青春期以后。
饮食治疗的目的是使血中苯丙氨酸保持在0.24~0.6mmol/L,患儿可以在低苯丙氨酸食品喂养的基础上,辅以母乳和牛奶。每100毫升母乳含苯丙氨酸约40mg,每30ml牛乳含50mg。限制苯丙氨酸摄入的特制食品价贵,操作起来有一定困难。至于饮食中限制苯丙氨酸摄入的饮食治疗,到何时可停止,迄今尚无统一意见,一般认为要坚持10年。在限制苯丙氨酸摄入饮食治疗的同时,联合补充酪氨酸或用补充酪氨酸取代饮食。饮食中补充酪氨酸可以使毛发色素脱失恢复正常,但对智力进步无作用。在限制苯丙氨酸摄入的饮食治疗过程中,应密切观察患儿的生长发育营养状况,及血中苯丙氨酸水平及副作用。副作用主要是其他营养缺乏,可出现腹泻、贫血(大细胞性)、低血糖低蛋白血症和烟酸缺乏样皮疹等。
2、药物治疗
2007年,Kuvan(盐酸沙丙蝶呤)被美国食品药品监督管理局(FDA)批准用于治疗PKU。 Kuvan 是 BH4 的口服药物制剂,BH4 是 PAH 酶的天然辅助因子,可刺激残留 PAH 酶的活性,将苯丙氨酸代谢为酪氨酸。 Kuvan 将与苯丙氨酸限制饮食一起使用。 Kuvan 由 BioMarin Pharmaceutical Inc. 制造。
2018年,Palynziq(pegvaliase-pqpz)被FDA批准用于成人PKU,2019年欧盟也批准其上市。 Palynziq 是一种可注射的酶疗法,适用于目前治疗中血液苯丙氨酸浓度不受控制的患者。 Palynziq 由 BioMarin Pharmaceutical Inc. 制造。见:欧盟批准Palynziq用于治疗苯丙酮尿症成年患者
3、基因治疗
FDA 停止了在 PKU 成人中研究其 AAV5-苯丙氨酸羟化酶 (PAH) 基因治疗候选药物 BMN 307 的 I/II 期 PHEARLESS 试验。
六、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
FDA暂停了苯丙酮尿症(PKU)基因疗法BMN 307的I/II期临床试验
De la Cruz F, Koch R. Genetic Implications for newborn screening for phenylketonuria. Clin Perinatol. 2001;28:419-24.
van Spronsen FJ, Smit PG, Koch R. Phenylketonuria: tyrosine beyond the phenylalanine diet. J Inherit Metab Dis. 2001;24:1-4.
Griffith P. Neuropsychological approaches to treatment policy issues in phenylketonuria. Eur J Pediatr. 2000;159:Suppl 2:S82-86, comment Eur J Pediatr. 2000;159:Suppl 2:S87-88.
Van Spronsen FJ, van Rijn M, Bekhof J, et al. Phenylketonuria: tyrosine supplementation in phenylalanine restricted diets. Am J Clin Nutr. 2001;73:153-57.
Muntau AC, Röschinger W, Habich M, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347:2122-32.
Seashore MR. Tetrahydrobiopterin and dietary restriction in mild phenylketonuria. N Engl J Med. 2002;347:2094-95.
American Academy of Pediatrics, Committee on Genetics. American Academy of Pediatrics:Maternal phenylketonuria. Pediatrics. 2001;107:427-28.
National Institute of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16-18, 2000. Pediatrics. 2001;108:972-82.
Rohr FJ, Munier AW, Levy HL. Acceptability of a new modular protein substitute for the dietary treatment of phenylketonuria. J Inherit Metab Dis. 2001;24:623-30.
Erlandsen H, Stevens RC. A structural hypothesis for BH4 responsiveness in patients with mild forms of hyperphenylalaninemia and phenyketonuria. J Inhab Metab Dis. 2001;24:213-30.
Kalsner LR, Rohr FJ, Strauss KA, et al. Tyrosine supplementation in phenylketonuria: diurnal blood tyrosine levels and presumptive brain influx of tyrosine and other large neutral amino acids. J Pediatr;2001;139:421-27.
McKusick VA, Ed. Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Entry Number; 261600. Last Edit Date: 11/01/2017. https://www.omim.org/entry/261600?search=261600&highlight=261600 Accessed May 29, 2019.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#流行病#
48
#苯丙酮尿症#确实不少见
77