
Nat Med:多组学方法绘制结直肠癌肿瘤、免疫和微生物综合图谱,揭示微生物组与免疫系统的相互作用
2024-04-29 测序中国 测序中国 发表于上海

该研究使用的多组学方法能够彻底检测分析结直肠癌免疫反应的分子特征,并揭示微生物组与免疫系统之间的相互作用。
近年来,虽然对原发性结肠癌的生物标志物已经进行了大量研究,但目前的临床指南仅依赖于肿瘤-淋巴结-转移分期和DNA错配修复(MMR)缺陷或微卫星不稳定性(MSI)的检测(除标准病理检测)来确定治疗建议。研究人员已经注意到,癌症基因组图谱(TCGA)的结直肠癌队列中基于基因表达的免疫反应、微生物图谱、肿瘤基质等与患者生存之间缺乏关联。
随着研究的深入,原发性结直肠癌的定量特征,包括癌细胞、免疫、基质或微生物性质的癌症,已被报道与临床结果显著相关,但是关于它们的相互作用如何影响患者结果仍了解有限。
为了剖析表型复杂性与结果的关系,近期,卡塔尔Sidra医学研究所的研究团队通过结合微生物组特征和免疫排斥常数(ICR),开发并验证了综合评分(mICRoScore),该评分可识别一组具有良好生存率的患者。研究团队对来自348名原发性结直肠癌患者的新鲜冷冻样本进行了全面基因组分析,包括肿瘤和匹配的健康结直肠组织的RNA测序、全外显子组测序、深层T细胞受体和16S细菌rRNA基因测序,并辅以肿瘤全基因组测序以进一步表征微生物组。该研究已发表在Nature Medicine上,题目为“An integrated tumor, immune and microbiome atlas of colon cancer”。

文章发表在Nature Medicine上
AC-ICAM概述
研究人员使用正交基因组平台,对未经系统治疗的组织学诊断为结肠癌患者的新鲜冷冻肿瘤样本和匹配的邻近健康结肠组织(肿瘤-正常对)进行分析。基于全外显子组测序(WES)、RNA-seq数据质量控制和纳入标准筛选,348名患者的基因组数据被保留并用于下游分析,中位随访时间为4.6年。研究团队将此资源命名为Sidra-LUMC AC-ICAM:免疫-癌症-微生物组相互作用的图谱和指南(图1)。
使用ICR进行分子分类
捕获连续癌症免疫监测的一系列模块化免疫基因标记,称为排斥免疫常数(ICR),研究团队将ICR优化浓缩为一个20基因panel,覆盖了不同癌症类型,包括黑色素瘤、膀胱癌、乳腺癌等。ICR还与乳腺癌等多种癌症类型的免疫治疗反应相关。
首先,研究人员对AC-ICAM队列进行了ICR特征验证,利用一种基于ICR基因的共聚类方法将队列分为三个集群/免疫亚型:ICR高(热肿瘤),ICR中和ICR低(冷肿瘤)(图1b)。研究人员表征了与共识分子亚型(CMS)相关的免疫倾向,这一种基于转录组的结肠癌分类。CMS类别包括CMS1/免疫,CMS2/规范,CMS3/代谢和CMS4/间充质。分析显示,在所有CMS亚型中,ICR评分与某些癌细胞通路呈负相关,仅在CMS4肿瘤中观察到与免疫抑制和基质相关通路呈正相关。
在所有CMS中,自然杀伤(NK)细胞和T细胞亚群的丰度在ICR高免疫亚型中最高,其他白细胞亚群的可变性更大(图1c)。ICR免疫亚型具有不同的OS和PFS,ICR从低到高逐渐增加(图1d),验证了ICR在结直肠癌中的预后作用。
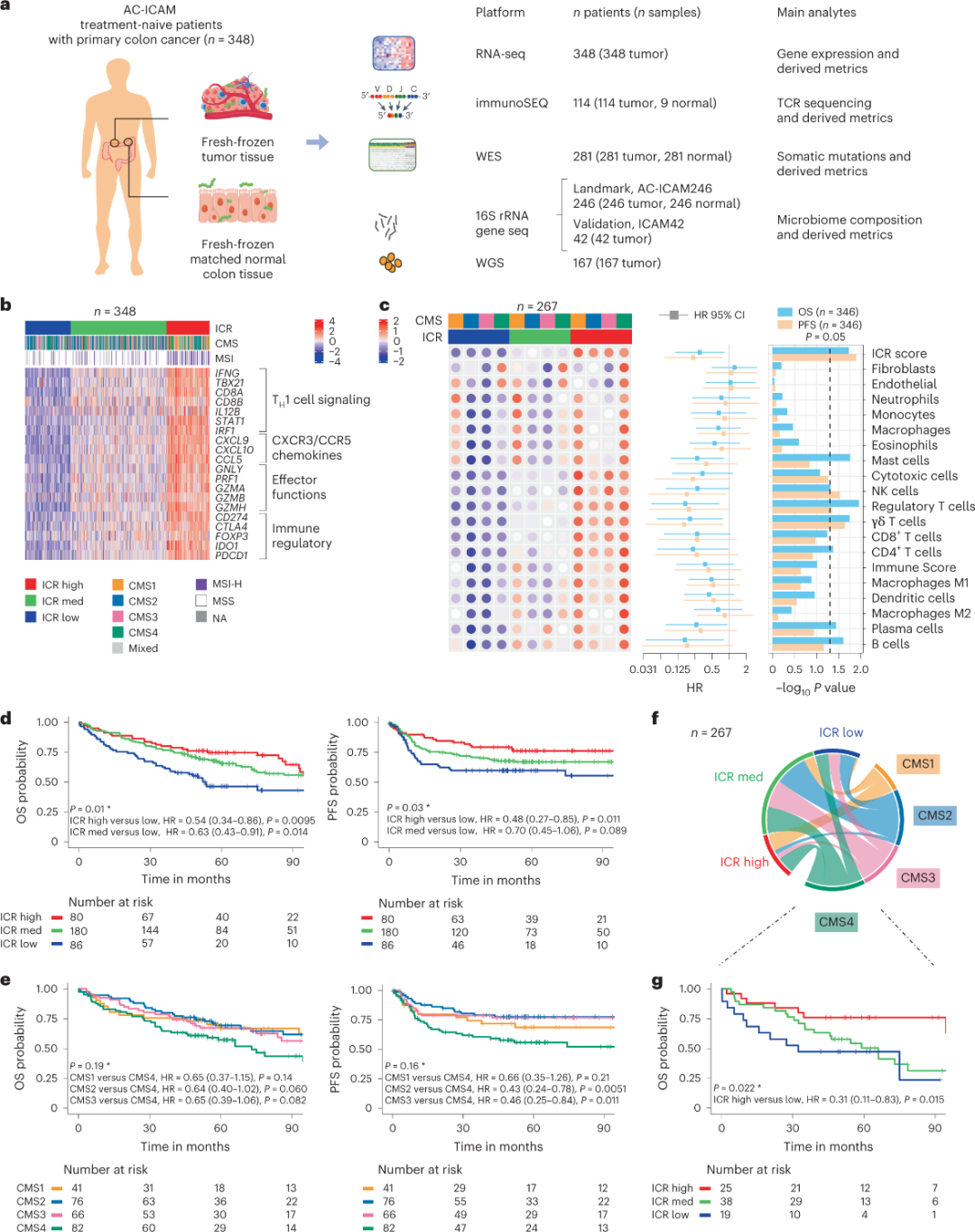
图1. AC-ICAM研究设计,免疫相关基因特征,免疫和分子亚型和生存期。
ICR捕获肿瘤富集、克隆扩增的T细胞
据报道,只有少数浸润肿瘤组织的T细胞对肿瘤抗原具有特异性(小于10%)。因此,大多数肿瘤内T细胞被称为旁观者T细胞(bystander T cells)。在基质细胞和白细胞亚群(通过RNA-seq检测)中,观察到了与具有 productive TCR的常规T细胞数量最强的相关性,可用于估计T细胞亚群(图2a)。在ICR簇(总体和CMS分类)中,ICR高组和CMS亚型CMS1/免疫组的免疫SEQ TCR克隆性最高(图2c),其中ICR-高肿瘤比例最高。使用全转录组(18270个基因),与TCR免疫SEQ克隆性正相关的前十个基因中有六个ICR基因(IFNG、STAT1、IRF1、CCL5、GZMA和CXCL10)(图2d)。与利用肿瘤反应性CD8+ 标记物观察到的相关性相比,免疫SEQ TCR克隆性与大多数ICR基因的相关性更强(图2f和2g)。总之,上述分析表明,ICR特征捕获了肿瘤富集、克隆扩增T细胞的存在,可能解释了其预后的含义。
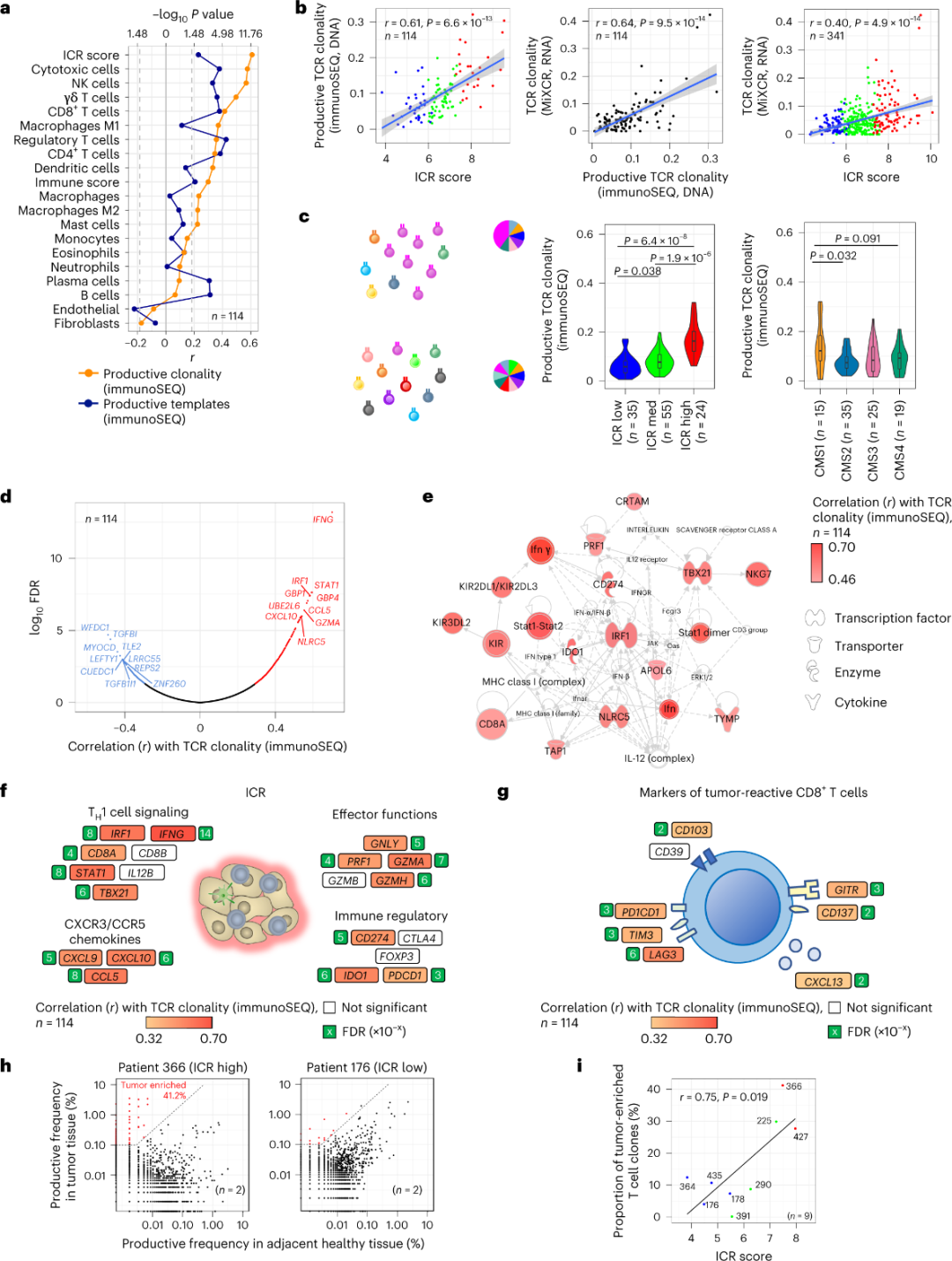
图2. TCR指标以及与免疫相关基因,免疫和分子亚型的相关性。
健康和结肠癌组织中的微生物组组成
研究人员利用从246名患者的匹配肿瘤和健康结肠组织中提取的DNA进行16S rRNA测序(图3a)。为了验证,研究人员额外分析了另外42个肿瘤样本的16S rRNA基因测序数据,这些样本没有匹配的正常DNA可用于分析。首先,研究人员比较了匹配的肿瘤和健康结肠组织之间菌群的相对丰度。与健康样本相比,肿瘤中的梭杆菌显著增加(图3a-3d)。肿瘤样本和健康样本的α多样性(单个样本中物种的多样性和丰度)没有显著差异,相对于ICR-低肿瘤,在ICR-高肿瘤中观察到适度减少的微生物多样性。
为了检测微生物谱和临床结果之间的临床相关联系,研究人员旨在使用16S rRNA基因测序数据来识别预测存活率的微生物组特征。在AC-ICAM246上,研究人员运行了一个OS Cox回归模型,该模型选择了41个系数不为零的特征(与差异死亡风险相关),称为MBR分类器(图3f)。
在该训练队列(ICAM246)中,低MBR评分(MBR<0,MBR低)与死亡风险显着降低(85%)相关。研究人员在两个独立验证队列(ICAM42和TCGA-COAD)中确认了MBR低(风险)和延长OS之间的关联。(图3)研究显示,内瘤胃球菌和MBR评分存在强相关性,两者在肿瘤和健康结肠组织中相似。
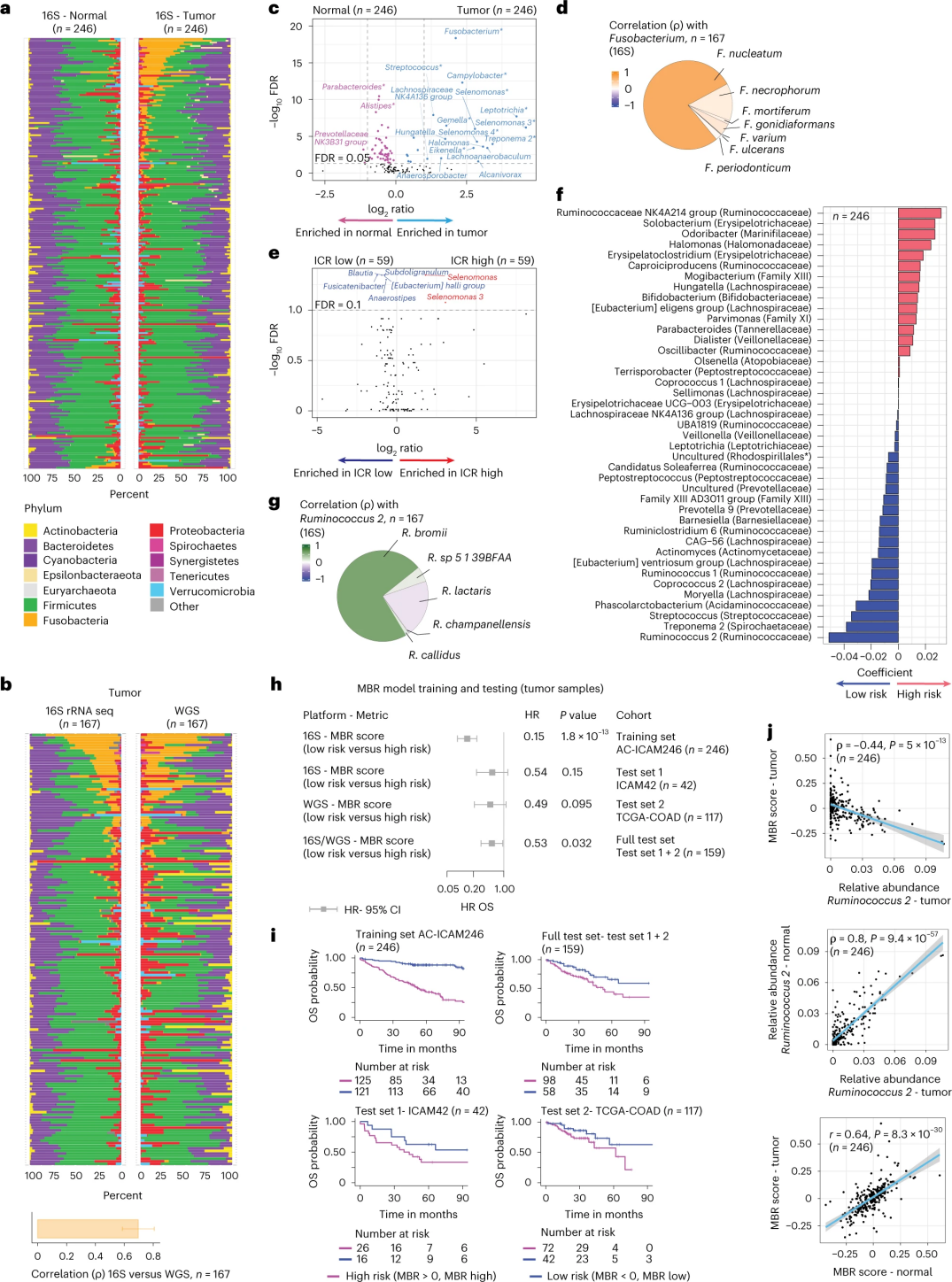
图3. 肿瘤和健康组织中的微生物组以及与ICR和患者生存的关系。
结 语
该研究使用的多组学方法能够彻底检测分析结直肠癌免疫反应的分子特征,并揭示微生物组与免疫系统之间的相互作用。对肿瘤和健康组织进行深度TCR测序,发现ICR的预后效果可能是由于其能够捕获肿瘤富集且可能具有肿瘤抗原特异性的T细胞克隆。
通过在AC-ICAM样本中使用16S rRNA基因测序分析肿瘤微生物组组成,研究团队确定了一个具有较强预后价值的微生物组特征(MBR风险评分)。虽然这一特征来自肿瘤样本,但健康结直肠与肿瘤MBR风险评分之间存在很强的相关性,表明该特征可能捕获了患者的肠道微生物组组成。通过结合ICR和MBR评分,能够识别和验证一种多组学生物标志物,该标志物可以预测结肠癌患者的生存期。该研究的多组学数据集为更好地理解结肠癌生物学提供了资源,有助于发现个性化的治疗方法。
参考资料:
Roelands, J., Kuppen, P.J.K., Ahmed, E.I. et al. An integrated tumor, immune and microbiome atlas of colon cancer. Nat Med 29, 1273–1286 (2023).
https://www.nature.com/articles/s41591-023-02324-5
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#结直肠癌肿瘤#
3